نقص ألفا-1 أنتي تريبسين: دليل شامل عن هذا المرض الوراثي الصامت الذي يدمر الرئتين والكبد، أسبابه الجينية في جين SERPINA1، أعراضه، تشخيصه، وأحدث علاجاته من التعويض البروتيني إلى CRISPR.
⚡ الإجابة السريعة
ما هو نقص ألفا-1 أنتي تريبسين؟
نقص ألفا-1 أنتي تريبسين (AATD) هو اضطراب وراثي ناتج عن طفرات في جين
SERPINA1، يؤدي إلى انخفاض شديد أو غياب بروتين “ألفا-1” الذي تصنعه الكبد
ومهمته حماية الرئتين من إنزيمات هاضمة تدمر أنسجتهما. يُعدّ من أكثر الأمراض الوراثية الخطيرة
شيوعاً لكنه الأقل تشخيصاً عالمياً — يتأخر تشخيصه في المتوسط
7-8 سنوات من ظهور الأعراض. يؤثر على الرئتين (نفاخ رئوي مبكر)
وعلى الكبد (تليف وسرطان).
| السبب | طفرة في جين SERPINA1 — أشيعها الأليل Z (نمط PiZZ) |
| التأثير على الرئة | نفاخ رئوي شامل مبكر — داء انسداد رئوي مزمن في سن مبكرة |
| التأثير على الكبد | تراكم البروتين المشوه في الخلايا الكبدية — تليف وسرطان كبد |
| التشخيص | قياس مستوى البروتين في الدم + اختبار جيني + أشعة مقطعية HRCT |
| العلاج الحالي | العلاج التعويضي البروتيني الوريدي — الوحيد المعتمد للرئتين |
| أقوى عامل خطر | التدخين — يقلص العمر المتوقع 10-15 سنة ويعجّل تدمير الرئة |
نقص ألفا-1 أنتي تريبسين (Alpha-1 Antitrypsin Deficiency) الدليل الشامل للأسباب والأعراض وطرق العلاج الحديثة
ما هو نقص ألفا-1 أنتي تريبسين؟
يعتبر نقص ألفا-1 أنتي تريبسين (AATD) اضطراباً وراثياً يتميز بانخفاض مستويات التداول أو خلل وظيفي لبروتين “ألفا-1 أنتيتريبسين”، وهو مثبط حيوي لإنزيمات البروتياز يُصنع أساساً بالكبد. وُصف لأول مرة عام 1963، ولا يزال أحد أكثر الحالات الوراثية الخطيرة شيوعاً عالمياً، ومع ذلك لا يُشخص بشكل كافٍ، مع تأخر تشخيصي يبلغ سبع إلى ثماني سنوات. يتجلى المرض أساساً بنفاخ رئوي تدريجي مبكر، ولدى فئة من المرضى، مرض كبدي مزمن يتراوح من التهاب الكبد الوليدي إلى تليف الكبد الخفي وسرطان الخلايا الكبدية.
التعرف على هذا النقص له آثار سريرية كبيرة، حيث إن التدخلات الموجهة، وتعديلات نمط الحياة، والعلاجات الخاصة بالمرض يمكن أن تغير مسار الحالة، وتحسن جودة الحياة، وتوجه فحص الأسرة. يقدم هذا المقال نظرة شاملة عن المرض، تشمل أساسه الجيني، والفيزيولوجيا المرضية الجزيئية، والمظاهر السريرية، وخوارزميات التشخيص، واستراتيجيات الإدارة الحالية، والابتكارات العلاجية الناشئة.
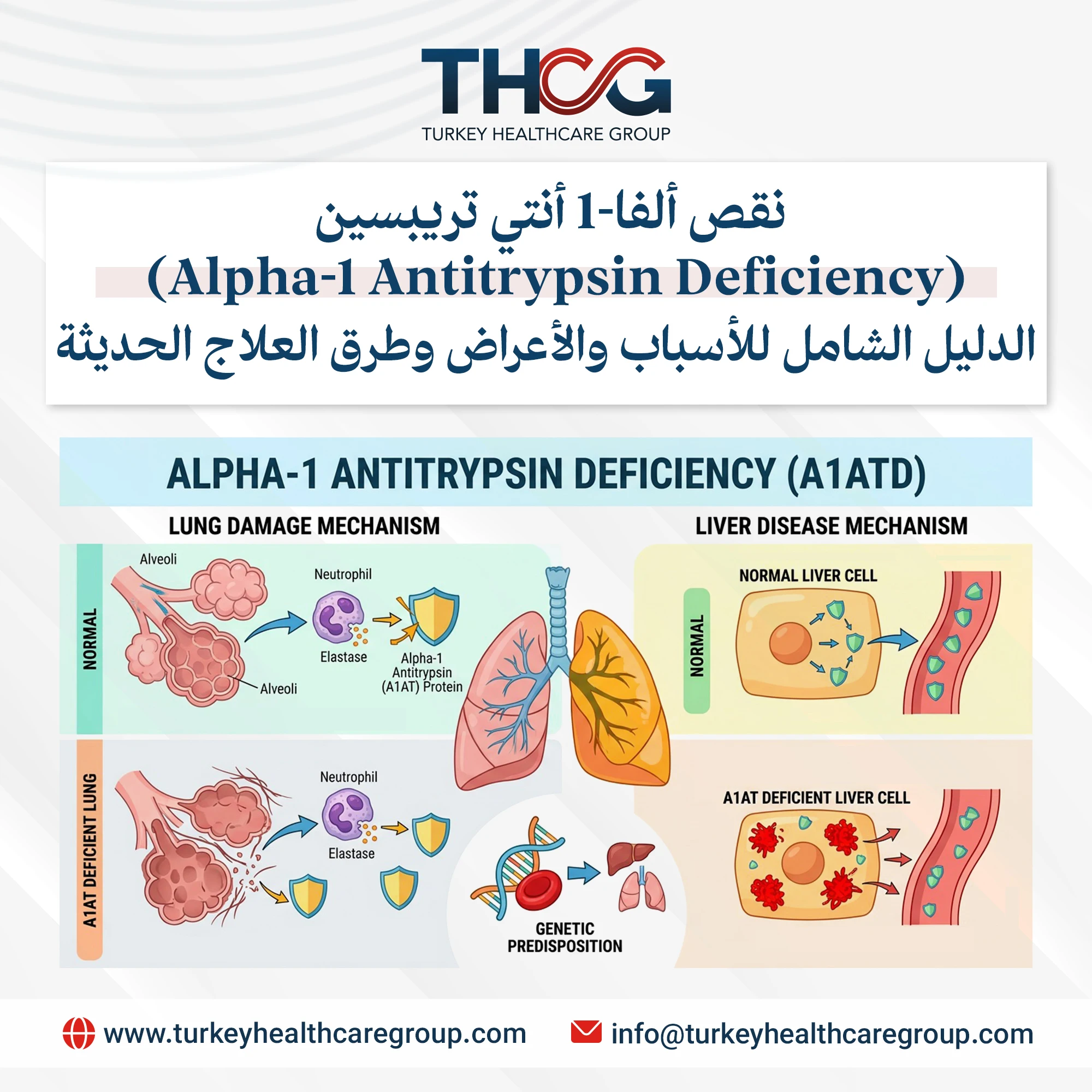
الوراثة والفيزيولوجيا المرضية الجزيئية
ينتج المرض عن طفرات جينية في جين (SERPINA1) الواقع على الكروموسوم 14. يشفر الجين بروتين “ألفا-1″، وهو بروتين سكري بوزن 52 كيلو دالتون يعمل كمثبط لإيلاستاز العدلات في الجهاز التنفسي السفلي. تم تحديد أكثر من 150 نوعاً من المتغيرات الجينية، لكن المرض السريري يقوده غالباً الأليل (S) والأليل (Z). النوع الطبيعي (M) ينتج بروتيناً وظيفياً بالكامل يفرز في مجرى الدم.
بينما يحفز الأليل (Z) تغيراً بنيوياً يعزز تراكم البروتين داخل الخلايا الكبدية. تؤدي آلية “كسب الوظيفة السام” هذه إلى إجهاد الخلايا الكبدية، وموتها المبرمج، والتهاب مزمن، مما يدفع في النهاية نحو التليف وتشمع الكبد لدى الأفراد المعرضين للإصابة.
بالتزامن، يؤدي جانب “فقدان الوظيفة” في هذا النقص إلى انخفاض ملحوظ في تركيزات المصل (عادة 10-15% من الطبيعي في أفراد PiZZ). بدون وجود كمية كافية لتحييد إيلاستاز العدلات وإنزيمات البروتياز الأخرى، يقوم النشاط البروتيني غير المنضبط بتكسير بروتينات الرئة، وخاصة الإيلاستين. تطلق هذه العملية التهاباً مزمناً، وإجهاداً تأكسدياً، وتدميراً متسارعاً لأنسجة الرئة، مما ينتهي بنفاخ رئوي شامل.
تسلط الأبحاث الناشئة الضوء أيضاً على أدوار أوسع لهذا البروتين في تنظيم المناعة، بما في ذلك حماية الأوعية الدموية والحواجز الخلوية، مما يشير إلى أن نقصه قد يمهد لمضاعفات التهابية وجهازية تتجاوز الأنماط الظاهرية الرئوية والكبدية الكلاسيكية المعروفة.
من هم الأكثر عرضة للإصابة؟
يختلف انتشار هذا النقص بشكل كبير حسب الموقع الجغرافي والأصل العرقي. يحدث النمط الظاهري (PiZZ) في حوالي 1 من كل 2,500 إلى 5,000 فرد من أصول أوروبية شمالية، مع ترددات عالية للناقلين من نوع (PiMZ). الانتشار أقل بكثير في الأصول الآسيوية والأفريقية، رغم أن ضعف التعرف ومحدودية الفحص يساهمان في بيانات وبائية غير مكتملة. عالمياً، يتم تشخيص أقل من 10% من المصابين بشكل صحيح، مما يمثل فجوة كبيرة في الصحة العامة.
تعدل عوامل البيئة ونمط الحياة التعبير عن المرض بشدة. تدخين السجائر هو أقوى مسرع للتدهور الرئوي، حيث يقلل متوسط العمر المتوقع بمقدار 10-15 سنة ويعجل بظهور النفاخ في سن مبكرة. كما يؤدي التعرض للغبار المهني، وتلوث الهواء، والعدوى التنفسية المتكررة، والربو غير المنضبط إلى تفاقم إصابة الرئة. وعلى العكس، قد يحافظ غير المدخنين المصابون بالمرض على وظائف رئوية شبه طبيعية حتى سن متأخرة، مما يؤكد التقاطع الحاسم بين الاستعداد الوراثي والمحفزات البيئية الخارجية.
ما هي أعراض المرض؟
يهيمن التأثير الرئوي والكبدي على الطيف السريري للمرض، رغم حدوث سمات جهازية أخرى.
المرض الرئوي: السمة المميزة هي داء الانسداد الرئوي المزمن المترقي والمبكر، مع نمط إشعاعي لنفاخ رئوي شامل يصيب أسفل الرئتين. وهذا يختلف عن النفاخ المرتبط بالتدخين الذي يصيب أعلى الرئة. يعاني المرضى من ضيق تنفس عند الجهد، وسعال مزمن، وأزيز، والتهاب شعب هوائية متكرر. قد يتطور توسع القصبات وارتفاع ضغط الدم الرئوي مع تقدم المرض. تظهر فئة من المرضى نمطاً شبيهاً بالربو، مما يؤدي غالباً لتشخيص خاطئ في البداية.
المرض الكبدي: ينبع تضرر الكبد من تراكم بروتينات (Z) المشوهة داخل الخلايا الكبدية. في المواليد، يعد هذا النقص سبباً رئيسياً لليرقان الوراثي، حيث يصاب 10% بضرر كبدي كبير. يعاني معظم الرضع من شفاء تلقائي، لكن البعض يتقدمون نحو التليف. في البالغين، يفسر النقص بعض حالات تليف الكبد الخفي ويزيد خطر الإصابة بسرطان الكبد، بغض النظر عن الفيروسات أو الكحول. وغالباً ما يظل مرض الكبد صامتاً حتى يتطور تليف متقدم.
مظاهر أخرى: يرتبط التهاب السبلة الشحمية الناخر، وهو حالة نادرة تظهر كعقيدات مؤلمة تحت الجلد، ارتباطاً وثيقاً بهذا النقص. وبشكل أقل شيوعاً، تم الإبلاغ عن التهابات أوعية دموية ومشاكل كلوية، رغم أن العلاقات السببية لا تزال قيد البحث.
ما هي طرق تشخيص المرض؟
- ضرورة الحفاظ على “مؤشر اشتباه عالٍ” لفحص أي شخص يعاني من:
- انسداد رئوي مزمن أو ربو لا يستجيب للعلاج التقليدي.
- توسع غير مبرر في الشعب الهوائية.
- أمراض كبد مجهولة السبب (بغض النظر عن العمر أو التدخين).
- الخطوة الأولى (فحص الدم): البدء بقياس كمية بروتين “ألفا-1” في الدم؛ حيث تشير المستويات المنخفضة بقوة إلى وجود النقص.
- ملاحظة: الالتهابات أو الحمل قد يرفعان النسبة مؤقتاً مما قد يضلل التشخيص.
- الاختبارات التأكيدية: إجراء فحص جيني دقيق وتنميط ظاهري لتحديد نوع الطفرة (مثل نظام Pi العالمي) ورصد السلالات النادرة.
- تقييم حالة الرئتين: ويشمل:
- اختبارات كفاءة التنفس وقدرة الرئة على تبادل الأكسجين.
- الأشعة المقطعية عالية الدقة (HRCT) لتحديد حجم التلف.
- تقييم حالة الكبد: ويشمل:
- تحاليل وظائف الكبد وفحص “مرونة الكبد” (الأشعة التداخلية).
- في حالات نادرة، يتم أخذ عينة من الكبد للتأكد من وجود حبيبات البروتين المتراكمة.
- فحص العائلة: التوصية بفحص الأقارب من الدرجة الأولى فور اكتشاف إصابة أي شخص، لأن التدخل المبكر يحسن النتائج الصحية بشكل جذري.
ما هي طرق العلاج الحديثة لمرض نقص ألفا-1 أنتي تريبسين؟
تعتمد إدارة نقص “ألفا-1 أنتيتريبسين” على نهج متعدد المحاور يستهدف السيطرة على الأعراض، والحد من التدهور الوظيفي للأعضاء، والتدخل النوعي لتعويض النقص البروتيني.
أولاً: العلاج التعويضي البروتيني (Augmentation Therapy)
يُعد العلاج التعويذي بالحقن الوريدي لبروتين “ألفا-1 أنتيتريبسين” المستخلص من البلازما البشرية التدخل النوعي الوحيد المعتمد حالياً لعلاج المظاهر الرئوية للمرض.
- الآلية الوظيفية: يهدف هذا العلاج إلى رفع مستويات البروتين في المصل فوق “عتبة الحماية” (Protective Threshold)، مما يعيد التوازن بين إنزيمات البروتياز ومثبطاتها داخل النسيج الرئوي.
- المؤشرات السريرية: يُنصح به بصفة أساسية للمرضى من ذوي الأنماط الظاهرية الشديدة (مثل PiZZ) الذين أظهروا تدهوراً مستمراً في وظائف الرئة (FEV1) أو دليلاً إشعاعياً على نفاخ رئوي مترقٍ.
- الأثر السريري: أظهرت الدراسات السريرية الموسعة قدرة هذا العلاج على إبطاء معدل فقدان كثافة النسيج الرئوي كما يظهر في صور الأشعة المقطعية الكمية.
ثانياً: الإدارة الدوائية والوقائية (التدبير الرئوي)
تتبع إدارة المظاهر التنفسية البروتوكولات القياسية لعلاج داء الانسداد الرئوي المزمن (COPD)، مع تشديد الرقابة الوقائية:
- العلاجات الاستنشاقية: استخدام موسعات الشعب الهوائية طويلة الأمد (LABA/LAMA) والكورتيكوستيرويدات الاستنشاقية لتحسين سعة التنفس وتقليل معدل نوبات التفاقم.
- التدخل الوقائي الصارم: يعتبر الإقلاع النهائي عن التدخين حجر الزاوية في العلاج؛ حيث إن التدخين يعمل على أكسدة بروتين AAT وتعطيله وظيفياً، مما يسرع دمار الأنسجة.
- التحصين المناعي: ضرورة الالتزام بلقاحات الإنفلونزا السنوية ولقاحات المكورات الرئوية لتقليل مخاطر العدوى التنفسية الحادة التي تثير النشاط الإنزيمي المدمر.
ثالثاً: التدخلات الجراحية
في الحالات المتقدمة التي لا تستجيب للعلاجات الدوائية، يتم اللجوء إلى الخيارات الجراحية:
- جراحات تقليص حجم الرئة (LVRS): لتحسين ميكانيكا التنفس في حالات مختارة من النفاخ الرئوي.
- زراعة الكبد (Liver Transplantation): تُعد علاجاً شافياً للمظاهر الكبدية والرئوية معاً في حالات الفشل الكبدي؛ إذ يقوم الكبد المزروع بإنتاج البروتين السليم وتصحيح العجز الكيميائي الحيوي في الجسم.
- زراعة الرئة: تُعتبر الخيار الأخير لمرضى الفشل التنفسي النهائي، مما يساهم في تحسين جودة الحياة ومعدلات البقاء.
رابعاً: العلاجات الجينية
تتركز البحوث الحالية على تطوير تقنيات “إسكات الجينات” (Gene Silencing) لتقليل إنتاج البروتين المشوه في الكبد، وتقنيات تحرير الجينات (CRISPR) لتصحيح الطفرة الوراثية من جذورها، مما يبشر بتحول جذري في مستقبل علاج هذا الاضطراب.
مستقبل الحالة الصحية والتوقعات طويلة الأمد
تختلف التوقعات المستقبلية لمصابي نقص “ألفا-1” بشكل كبير من شخص لآخر، حيث يتحكم في هذا المسار ثلاثة عوامل رئيسية: التدخين، والنوع الجيني للطفرة، ومدى جودة الرعاية الطبية المستلمة. بالنسبة للأشخاص الذين لا يدخنون، حتى من لديهم النمط الجيني الحاد (PiZZ)، فغالباً ما يعيشون حياة طبيعية ومستقلة حتى سن السبعين أو الثمانين. وفي المقابل، يواجه المدخنون خطراً حقيقياً يتمثل في تدهور وظائف الرئة بشكل متسارع، مع التعرض لنوبات تنفسية حادة ومتكررة قد تؤدي -لا قدر الله- إلى فشل تنفسي أو تأثر عضلة القلب في سن مبكرة.
أما فيما يخص صحة الكبد، فإن أغلب الأطفال الذين يولدون بمشاكل كبدية مرتبطة بهذا النقص يتجاوزون هذه المرحلة بسلام مع مرور الوقت، لكن فئة قليلة منهم قد تحتاج لعملية زراعة كبد نتيجة تليف متقدم. وبالنسبة للبالغين، فإن التليف الكبدي يحمل معه المخاطر المعتادة لأمراض الكبد المزمنة، مثل احتمالية حدوث نزيف معوي أو أورام كبدية، مما يتطلب مراقبة طبية دقيقة.
لا تقتصر تأثيرات المرض على الجانب العضوي فحسب، بل تمتد لتؤثر على جودة الحياة اليومية؛ فصعوبة التنفس المزمنة قد تؤدي للشعور بالقلق أو الاكتئاب، بالإضافة إلى عبء المراجعات الطبية المستمرة. ومع ذلك، تظل الصورة متفائلة؛ إذ إن التشخيص المبكر والالتزام بالعلاجات التعويضية والمتابعة الدورية مع فريق طبي متكامل (يضم أطباء الرئة والكبد والوراثة والتغذية) يساهم بشكل جذري في تحسين مسار المرض، ويضمن للمصابين حياة أكثر استقراراً وجودة على المدى الطويل.
العلاجات الناشئة والتوجهات المستقبلية
يشهد المشهد العلاجي لنقص AATD تحولاً سريعاً. أظهرت وكلاء “الرنا المتدخل الصغير” (siRNA)، ولا سيما عقار (ARO-AAT)، قدرة قوية على خفض مستويات بروتين (Z-AAT) المتحور في تجارب المرحلة الثانية، مع استمرار التثبيط لمدة تصل لستة أشهر بعد جرعة واحدة. وتقوم برامج المرحلة الثالثة حالياً بتقييم السلامة والنتائج السريرية، مع توقع الموافقة عليها في السنوات القادمة. كما تتقدم تقنيات تحرير الجينات باستخدام (CRISPR-Cas9) لتعطيل جين (SERPINA1) أو تصحيح طفرة (Z) في الخلايا الكبدية عبر الدراسات قبل السريرية والبشرية المبكرة، مما يفتح آفاقاً لتدخل علاجي شافٍ لمرة واحدة.
يتم أيضاً تطوير بروتين (AAT) معاد التركيب، وتركيبات مستنشقة تستهدف الشعب الهوائية البعيدة مباشرة، وجزيئات صغيرة مصممة لتحسين طي البروتين وتسهيل خروجه من الكبد. تهدف معززات “البلعمة الذاتية” ومنظمات التوازن البروتيني إلى تسريع تنظيف الكبد من البوليمرات المتراكمة. بالتزامن،
يتم التحقق من صحة تحليل الأشعة المقطعية المعتمد على الذكاء الاصطناعي والمؤشرات الحيوية السائلة للتنبؤ بتقدم المرض وتخصيص جرعات العلاج التعويضي. إن تعزيز فحص حديثي الولادة، وتعميم اختبارات الانسداد الرئوي والربو، ومبادرات السجلات العالمية ستؤدي لتقليل تأخير التشخيص وتمكين الإدارة الدقيقة.
نقص “ألفا-1 أنتيتريبسين” هو اضطراب جهازي محدد وراثياً وله مظاهر رئوية وكبدية عميقة. ورغم انتشاره، لا يزال نقص التشخيص عائقاً حاسماً أمام الرعاية المثلى. تعتمد الفيزيولوجيا المرضية على آلية مزدوجة من تراكم البوليمرات السامة في الكبد ونشاط الإنزيمات غير المنضبط في الرئتين.
يشكل التعرف في الوقت المناسب، والتعديل البيئي الصارم، والعلاج التعويضي القائم على الأدلة، والإدارة الاستباقية للمضاعفات أساس العلاج المعاصر. ومع وجود خط إنتاج قوي لعلاجات الرنا، وتحرير الجينات، والجزيئات الصغيرة المستهدفة، فإن آفاق إدارة هذا النقص تزداد تفاؤلاً. يجب على الأطباء الحفاظ على عتبة منخفضة للاختبار، ودعم التدخلات الموجهة بالخطوط الإرشادية، ودمج المرضى في مسارات الرعاية الشاملة لتغيير التاريخ الطبيعي لهذه الحالة التي عانت تاريخياً من نقص التعرف عليها.
الأسئلة الأكثر بحثاً
نقص ألفا-1 أنتي تريبسين — كل أسئلتك بإجابات واضحة
إجابات دقيقة وموثوقة على أكثر الأسئلة شيوعاً حول هذا المرض الوراثي الصامت —
من التشخيص والأعراض إلى أحدث العلاجات وفرص الشفاء الجيني.
س1: لماذا يُسمى نقص ألفا-1 أنتي تريبسين “المرض الوراثي الصامت”؟
ج: لأن أعراضه الرئوية والكبدية تظهر ببطء شديد وتشبه أمراضاً شائعة أخرى كالربو وداء الانسداد الرئوي المزمن. يُشخَّص أقل من 10% من المصابين به عالمياً بشكل صحيح، ويتأخر التشخيص في المتوسط 7-8 سنوات من ظهور الأعراض الأولى، مما يسمح للضرر بالتراكم دون رادع.
س2: ما دور بروتين ألفا-1 أنتي تريبسين في الجسم وماذا يحدث عند نقصه؟
ج: وظيفته الأساسية حماية أنسجة الرئة من إنزيم “إيلاستاز العدلات” الذي يفرزه الجهاز المناعي لمحاربة العدوى. عند نقص البروتين، يعمل هذا الإنزيم بلا قيود فيهضم بروتين الإيلاستين في الرئة مسبباً التهاباً مزمناً وتدميراً تدريجياً لأنسجتها حتى يصل للنفاخ الرئوي الشامل.
س3: ما الفرق بين النفاخ الرئوي في هذا المرض والنفاخ الناتج عن التدخين؟
ج: الفرق في موقع الإصابة داخل الرئة. في نقص AATD يصيب النفاخ أسفل الرئتين بشكل رئيسي (Panlobular Emphysema)، بينما نفاخ التدخين يصيب أعلى الرئة. هذا الفارق الإشعاعي في الأشعة المقطعية HRCT يُنبّه الطبيب للبحث عن نقص ألفا-1 حتى في غير المدخنين.
س4: كيف يدمر نقص ألفا-1 الكبد إذا كان المشكلة في الرئة؟
ج: المشكلة مزدوجة. في الرئة: نقص البروتين يتركها بلا حماية. أما في الكبد: فالطفرة Z تجعل البروتين يتشوه ويتراكم داخل الخلايا الكبدية بدلاً من إفرازه للدم. هذا التراكم يُسبّب إجهاداً وموتاً للخلايا الكبدية وتليفاً تدريجياً وخطر سرطان الكبد — بمعزل تام عن الكحول أو الفيروسات.
س5: ما الأليلات الجينية الرئيسية وأيها الأخطر؟
ج: الأليل الطبيعي M ينتج بروتيناً وظيفياً كاملاً. الأليل Z هو الأخطر — يسبب تشوه البروتين وتراكمه في الكبد. الأليل S أقل خطورة. النمط PiZZ (أليلان Z من كلا الوالدين) هو الأشد حدة ويُنتج مستويات بروتين تبلغ 10-15% فقط من الطبيعي، وهو المرتبط بالمرض الرئوي والكبدي الشديد.
س6: هل يمكن للشخص المصاب بالمرض ألا تظهر عليه أعراض طوال حياته؟
ج: نعم، خاصةً غير المدخنين. من يملك النمط PiZZ ولا يدخن وتجنّب التعرض المهني للغبار وعاش في بيئة نظيفة، قد يحافظ على وظائف رئوية شبه طبيعية حتى السبعينيات والثمانينيات. هذا يُثبت أن البيئة تلعب دوراً محورياً يعادل الاستعداد الوراثي في التحكم بمسار المرض.
س7: كيف يؤثر التدخين تحديداً على مرضى نقص ألفا-1؟
ج: تأثيره مضاعف ومدمر. أولاً يحفز إفراز المزيد من إيلاستاز العدلات. ثانياً يُؤكسد بروتين ألفا-1 الموجود أصلاً بكميات شحيحة ويعطله وظيفياً، فيفقد الرئة حتى هذه الحماية الضئيلة. المحصلة: تدهور متسارع لوظائف الرئة يقلص العمر المتوقع 10-15 سنة ويُظهر النفاخ في سن مبكرة بشكل لافت.
س8: ما هو العلاج التعويضي البروتيني وكيف يُعطى؟
ج: هو حقن وريدي أسبوعي لبروتين ألفا-1 مستخلص من بلازما متبرعين بشريين أصحاء. يهدف لرفع مستوى البروتين في الدم فوق “عتبة الحماية” لاستعادة التوازن بين الإنزيمات ومثبطاتها في الرئة. هو العلاج النوعي الوحيد المعتمد حالياً للمظاهر الرئوية، ويُبطئ فقدان كثافة النسيج الرئوي لكنه لا يعالج الكبد.
س9: هل العلاج التعويضي البروتيني يُعالج الكبد أيضاً؟
ج: لا. العلاج التعويضي يُعوّض النقص في الدم ويحمي الرئة، لكن مشكلة الكبد مختلفة تماماً — هي تراكم البروتين المشوه داخل الخلايا الكبدية. الحل الوحيد لمشكلة الكبد المتقدمة هو زراعة الكبد التي تُنتج بروتيناً سليماً وتُصحّح العجز الكيميائي في آنٍ واحد.
س10: هل يُشخَّص نقص ألفا-1 في مسح حديثي الولادة؟
ج: في بعض الدول نعم، لكن ليس في معظم دول العالم. حيث يُطبَّق، يسمح الفحص المبكر بالتدخل الوقائي قبل ظهور أي ضرر. الفحص الأشمل هو قياس مستوى البروتين في دم الوليد. غياب هذا الفحص في كثير من المناطق هو المسبب الرئيسي للتأخر التشخيصي 7-8 سنوات.
س11: ما الحالات التي يجب فيها الاشتباه بنقص ألفا-1 والإسراع في الفحص؟
ج: يجب الفحص الفوري عند: داء انسداد رئوي مزمن في شخص أقل من 45 سنة أو غير مدخن، ربو لا يستجيب للعلاج التقليدي، نفاخ رئوي يصيب أسفل الرئة، توسع قصبات غير مبرر، أمراض كبد مجهولة السبب في أي عمر، أو وجود قريب مشخص بالمرض.
س12: لماذا قد تكون نتيجة فحص الدم مضللة في بعض الحالات؟
ج: لأن بروتين ألفا-1 من بروتينات المرحلة الحادة — يرتفع طبيعياً أثناء الالتهابات والعدوى والحمل وحتى التدخين. فقد يبدو المستوى “طبيعياً” في الدم رغم وجود النقص الجيني. لهذا يجب إجراء الفحص في حالة استقرار صحي، وتأكيد النتيجة بالاختبار الجيني.
س13: ما نظام Pi لتصنيف الأنماط الجينية وما أهميته السريرية؟
ج: هو نظام دولي لتسمية أنماط بروتين ألفا-1 حسب قدرته على التنقل الكهربائي. PiMM = طبيعي، PiZZ = أشد الحالات (10-15% من الطبيعي)، PiMZ = ناقل مع مستوى متوسط، PiSZ = متوسط الشدة. تحديد النمط بدقة يوجّه قرار العلاج التعويضي ومدى أهلية المريض له.
س14: كيف يختلف علاج نقص ألفا-1 عند الأطفال عن البالغين؟
ج: عند الأطفال تهيمن مشكلة الكبد (يرقان وليدي، تليف مبكر) وتكون الرئة سليمة في الغالب. معظم الأطفال يتجاوزون اليرقان تلقائياً لكن فئة تحتاج زراعة كبد مبكرة. عند البالغين تبرز المشكلة الرئوية تدريجياً. العلاج التعويضي البروتيني مخصص للبالغين ذوي التدهور الرئوي الموثق.
س15: ما عقار ARO-AAT وما أهميته في مستقبل علاج هذا المرض؟
ج: هو دواء من فئة “الرنا المتدخل الصغير” (siRNA) يعمل على إسكات جين SERPINA1 في الكبد لمنع إنتاج البروتين المشوه Z-AAT الذي يتراكم ويدمر الخلايا الكبدية. أظهر في تجارب المرحلة الثانية قدرة على تخفيض البروتين المتراكم بشكل كبير لمدة 6 أشهر بجرعة واحدة. تجارب المرحلة الثالثة جارية حالياً.
س16: ما الفرق بين زراعة الكبد وزراعة الرئة في علاج نقص ألفا-1؟
ج: زراعة الكبد تُعالج المشكلتين معاً — الكبد المزروع ينتج بروتيناً سليماً يحمي الرئة أيضاً، وهي بهذا المعنى علاج شافٍ للعجز الجيني الكيميائي. أما زراعة الرئة فتُستخدم عند فشل الرئة النهائي وتُحسّن جودة الحياة، لكنها لا تُصلح الخلل الجيني الأصلي في الكبد.
س17: هل فحص أفراد العائلة ضروري عند اكتشاف الإصابة؟
ج: نعم وبشكل عاجل. المرض وراثي متنحٍ يتوارثه الأشقاء والأبناء. فحص الأقارب من الدرجة الأولى فور التشخيص يكشف حالات في مراحل مبكرة قبل ظهور أعراض، مما يُتيح التدخل الوقائي بالإقلاع عن التدخين والعلاج المبكر — وهذا يُغير مسار المرض جذرياً.
س18: ما التطعيمات الضرورية لمريض نقص ألفا-1؟
ج: ضرورية وغير قابلة للتأجيل: لقاح الإنفلونزا السنوي، لقاح المكورات الرئوية (Pneumococcal)، لقاح التهاب الكبد A والتهاب الكبد B. أي عدوى تنفسية تُشعل استجابة مناعية تُفرز المزيد من إيلاستاز العدلات، مما يُسرّع تدمير أنسجة الرئة المحرومة أصلاً من الحماية.
س19: هل يمكن لمريض نقص ألفا-1 ممارسة الرياضة والحياة الطبيعية؟
ج: نعم، وهي مُشجَّعة بقوة. التمرين يُحسّن وظائف الرئة ويُبطئ التدهور. غير المدخنين حتى من حاملي النمط PiZZ يمكنهم العيش بشكل مستقل حتى السبعينيات. التحدي الأكبر هو التكيف مع جلسات العلاج التعويضي الأسبوعية وزيارات المتابعة، لكن مع التخطيط الجيد يبقى الأثر على جودة الحياة محدوداً.
س20: ما التوقعات المستقبلية لمرضى نقص ألفا-1 مع تطور العلاجات الجينية؟
ج: واعدة جداً. تقنية CRISPR-Cas9 تسعى لتصحيح طفرة Z في الخلايا الكبدية مرة واحدة للأبد. تقنيات siRNA كـ ARO-AAT تُوقف إنتاج البروتين المشوه. جزيئات صغيرة تُساعد البروتين على الطي الصحيح والخروج من الكبد. هذه العلاجات مجتمعة تُشير إلى إمكانية علاج حقيقي يُنهي الحاجة للعلاج التعويضي الأسبوعي مدى الحياة.